网站导航




摘 要: 鞘翅目害虫是目前档案库房中最常见的有害生物之一,害虫严重爆发时,被蛀食的档案纸张被大面积毁损且无法修复,所以档案虫情监测和预防极为重要。由于库房中害虫成虫采集难度大、害虫形态虫态差别大、发生规律各异、专业人员紧缺等原因,档案库房鞘翅目害虫的快速准确鉴定成为一个难点,为了更好地解决这个问题,本次研究我们利用DNA条形码鉴定技术将采集到的样本进行分子鉴定,利用线粒体基因组的COI基因片段,建立了档案鞘翅目害虫DNA条形码数据库。除此之外,本次研究中利用分子数据将3种害虫的成、幼虫配对,为解决幼虫形态鉴定困难的问题提供了新的思路。最后,我们讨论了如何更好地结合形态数据和DNA条形码数据进行物种鉴定,说明了DNA条形码技术能利用最少的取样实现快速高效鉴定档案鞘翅目害虫常见种类各虫态样本的目标。
关键词:档案害虫;DNA条形码;COI基因
DNA Barcoding Identification of Archives Coleoptera Pests
Abstract: Coleopteran are one of the most common pests in the archive rooms. In the case of serious pest outbreaks, the archival papers eaten by pests are irreparably damaged,so it is really important to monitor the situation and prevent the burst of pests in archives. The rapid and accurate identification of coleopteran pests has become a hard case due to the difficulty of collecting pests, the obvious individual differences, the disordered occurrence rules, and the shortage of professional taxonomist. In order to solve this problem, we used DNA barcoding technology to identify the archival pest samples based on the mitochondrial COI gene. Finally we established the archival coleopteran pest DNA barcode database including 9 coleoperan species and 4 other pests. Beyond that, this study found 3 pairings between adult and larva, which may provide a new way to solve the problem of identifying larva. At the work report of this study,we discussed how to better combine morphological data and DNA barcode data to identity species, we illustrated that DNA barcoding technology is a qualified method that can help researchers to identify archival coleopteran pests.
Key words: archival pests, DNA barcoding, COI gene
档案害虫属于仓储物害虫的一部分,目前我国有记载的仓储物害虫约500种[1],其中危害档案的害虫已发现71种。档案害虫分属于6目21科,其中鞘翅目昆虫共50种,是最常见的也是危害最严重的档案库房害虫[2] [3]。鞘翅目害虫多以蛀孔的方式对纸质档案造成破坏,受害严重的档案纸张被大面积损毁,无法补救,也会给国家档案保护和文化遗产保护工作造成严重的影响,因此对鞘翅目档案害虫的预防与治理工作是非常重要的。
种类准确识别鉴定是害虫防治的基础,鞘翅目昆虫已知物种数量近四十万,其分类鉴定工作以成虫形态研究为主,幼虫形态相似度高,较难鉴定。由于形态研究对鉴定人员的分类知识和经验要求较高,对害虫标本的保存情况要求也较高,实际工作中种类鉴定困难较大。近年来,快速发展的分子生物学技术为昆虫种类的快速高效鉴定提供了新的方法,其中DNA条形码分子鉴定技术是物种鉴定最有效的方法之一[4] [5],成熟的DNA条形码鉴定体系可以实现不同虫态、虫尸、碎片的快速准确物种鉴定[6] 。然而,档案害虫分子生物学研究仍处于初级阶段,且目前的仓储物害虫分子研究多集中于储粮害虫和烟草制品害虫[7] ,如危害储粮安全的拟步甲科[8] 、皮蠹科[9] [10]害虫等,并没有针对档案害虫的DNA条形码鉴定研究。
因此,本次开展鞘翅目档案害虫DNA条形码研究,首先是为了填补档案保护工作中害虫分子鉴定技术的空白,有效的弥补传统形态分类鉴定方法的短板,提高鉴定的准确度和效率。同时,研究成果可以为其他行业生物分子鉴定研究提供理论和技术参考。
DNA序列由A、T、G、C四种碱基组成,如果有n个碱基,就会有4n种编码方式。按照这个公式计算,15个碱基位点就能出现近10亿种的编码序列,这个数字是现存物种的100倍,再考虑到在蛋白编码基因中密码子的简并性,那么也只需要有45个碱基位点,就可以获得近10亿个DNA序列。由此看来,DNA条形码可以建立在一段长度为几百个碱基的基因序列信息的基础之上,从理论上来讲完全可以包括所有物种。这为DNA条形码分类工作的可实现性提供了条件,但是生物的基因组往往含有数万个甚至数十万个碱基位点,所以选择哪一部分基因作为标记就成为了另一个问题。理想的DNA条形码应该符合以下几个标准:(1)在种间有明显的遗传变异和分化,同时种内变异足够小;(2)片段足够短,便于完成测序工作,而且便于DNA提取和PCR扩增,尤其是对存在DNA降解的材料(如:保存已久的浸泡标本、处理过的民间药材);(3)存在保守区域,便于设计通用引物。也就是说这样的标记既要有一点的保守性又要有一定的变异性,片段的长度也要适中,而且还需要考虑到假基因和基因插入或缺失的情况(见下文)。从目前的研究结果来看,动物的DNA条形码标记选择趋向于COI基因,原核生物的标记则趋向于16S rRNA基因,对植物而言,人们提出过以rbcLa基因和matK基因作为标记,但是都受到了争议 [11],真菌界条形码基因的选择则仍在探寻阶段[12]。
尽管存在很多怀疑和反对,DNA条形码技术的发展依旧迅速,至2021年底,生命条形码数据系统已经有超过一千万个DNA条形码数据。生命条形码数据系统中的绝大多数条形码来自动物(超过230万个物种的数据),其次是植物(约7万种植物为rbcL序列)和真菌(2万4千多物种为ITS序列),而多种多样的单细胞原核生物仍然有很多未被发掘。
2.1 实验思路
(1)研究样本选取与标本收集
基于“全国档案害虫种类及分布调查”项目的调研结果以及目前的标本积累情况和文献资料调研,确定本研究所选取的鞘翅目档案害虫种类。根据研究需要制定标本采集计划并开展标本收集工作。研究材料主要基于目前已有的馆藏标本及饲养实验昆虫,并通过利用粘虫板等昆虫采集方法,补充收集用于DNA条形码研究的新鲜标本。同时,向其他开展仓储物害虫研究的单位借用定名标本,补充标本采集空白。
(2)形态分类鉴定
利用形态分类方法进行物种鉴定,除根据文献进行鉴定外,尽可能核对模式或定名标本,完成成虫拍照及其图片处理。
(3)DNA提取和聚合酶链式反应(Polymerase Chain Reaction,PCR)扩增方案制定
通过对标本的预处理试验和对DNA提取实验步骤的优化,制定鞘翅目档案害虫DNA提取的具体方案,确保DNA提取的有效浓度和纯度;
通过对PCR体系的优化和PCR程序的调试确定该类群DNA条形码的最适PCR条件;
在最优的DNA提取和PCR扩增方案基础上,对所有采样进行DNA模板的提取和条形码片段的扩增。
(4)筛选最适DNA条形码标记
根据已有的档案库房鞘翅目害虫名录,从国际通用数据库中查询相关DNA条形码数据,基于对COI、28S、18SrRNA三种常见基因分子标记的国际通用引物的修饰改进,确定不同分子标记引物的扩增效率,调查最适用于鞘翅目档案害虫的DNA条形码分子标记。
(5)档案鞘翅目害虫种间遗传距离估算
整理汇总查询的DNA条形码数据和本项目补充的条形码数据,估算档案库房鞘翅目害虫DNA条形码片段的种间遗传距离和种内遗传距离,确定种间遗传距离足够作为分子鉴定依据。
(6)基于分子数据的系统发育分析
利用筛选出的最适DNA条形码分子标记,可采用最大简约法,最大似然法,或者邻接法三种方法构建系统发育树。
(7)DNA条形码体系的建立
基于下载的与档案库房鞘翅目害虫近缘类群的DNA条形码数据,以及本研究室采集到的DNA条形码数据,分析物种间和物种内的遗传距离差异,确保DNA条形码对物种界定的有效性,利用系统发育树、barcoding gap分析,对筛选出的最适宜分子标记进行分析评价,判断其作为DNA条形码的有效性,确定其可否达到物种鉴定目的。
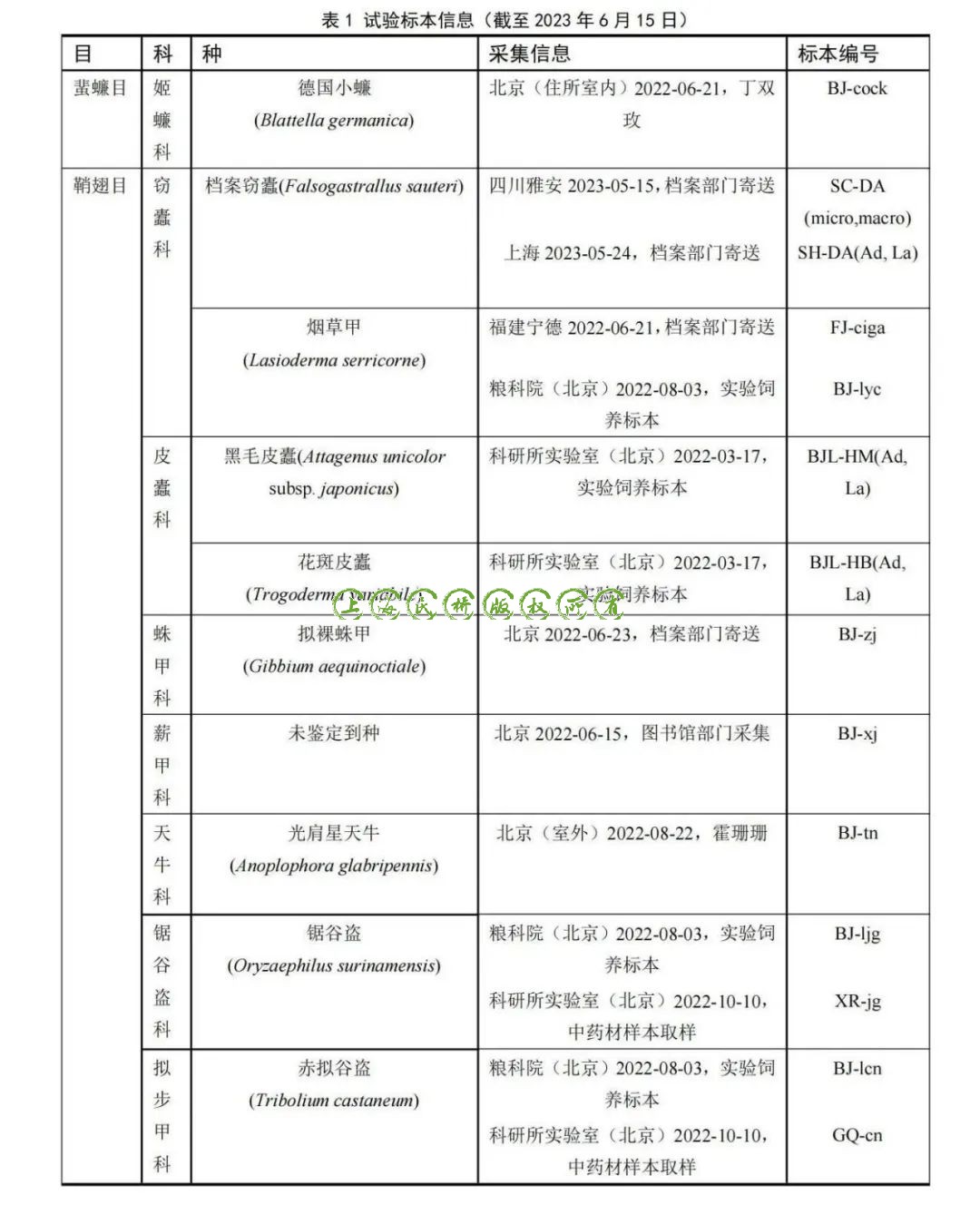
本试验所收集的标本信息见表1。所有样本采集后保存在95%的酒精中,-10℃保存备用。
主要实验仪器包括:离心机,恒温水浴锅,PCR仪,水平式电泳槽,稳压稳流电泳仪,紫外凝胶成像仪,组织研磨器。
主要化学试剂包括:血液/细胞/组织基因组DNA提取试剂盒(离心柱型)、微量样品基因组DNA提取试剂盒(离心柱型)、Taq Plus DNA Polymerase(500,2.5U/μL)、2×Taq PCR MasterMix II(带核酸染剂)、蛋白酶K、2.5mM dNTPs、10×Taq 缓冲液、6×DNA上样缓冲液、Gold View核酸染料、琼脂糖、D2000 DNA Maker、引物。
使用“血液/细胞/组织基因组DNA提取试剂盒(离心柱型)”进行基因组DNA的提取,具体操作参考试剂盒说明书。样品预处理步骤为:取液浸(无水乙醇)标本,成虫取整个胸部,幼虫去掉头部取剩余部位,单头样本放入2ml离心管中。对于较小的样品,可取2~3头,放入离心管后用组织研磨器研磨粉碎。
对保存时间超过半年、虫态为幼虫、单头标本体长不超过2mm的样本,使用天“微量样品基因组DNA提取试剂盒(离心柱型)”进行基因组DNA的提取,具体操作参考试剂盒说明书。样品预处理步骤为:取液浸(无水乙醇)标本,晾干酒精,体长超过2mm的成虫取整个胸部,幼虫去掉头部取剩余部位,单头样本放入2ml离心管中。对于较小的样品,可取2-3头,放入离心管后用组织研磨器研磨粉碎。
PCR反应的扩增体系为25μl,其中包括去离子水 14.5μl、10×Taq 缓冲液 5.0μl、2.5mM dNTPs 2.0μl、上游引物1.0μl、下游引物1.0μl、DNA模板1.0μl、2.5U/μl的Taq聚合酶0.5μl。
如果使用2×Taq PCR MasterMix II(带核酸染剂),则25μl体系为:2×Taq PCR MasterMix II 13.0μl、ddH2O 9.0μl、上游引物1.0μl、下游引物1.0μl、DNA模板1.0μl。
PCR反应条件为94℃预变性1分钟(模板裂解严重的缩短为30秒),94℃变性30秒(模板裂解严重的缩短为10秒)、43℃退火45秒(退火温度因引物而异,此处为COI基因片段退火温度,核基因片段一般为48℃或更高)、65℃延伸1分钟进行35个循环,最后65℃延伸10分钟后,5℃保存。
取5μl 6×DNA 上样缓冲液,与扩增的样品基因组DNA原液混匀,上样于1.5%琼脂糖凝胶中(如果使用Taq PCR MasterMix为扩增体系,则直接从扩增产物原液中取5μl上样),以1×TAE为电泳缓冲液,以D2000 DNA分子量标准作为分子量的标准对照,在220V稳定电压下电泳约20min,直至指示剂距低端约1cm。在紫外凝胶成像仪下观察检测提取的DNA片段的质量。
经电泳检验合格的PCR产物交由生工生物工程(上海)有限公司进行双向测序。使用Conting Express1软件检查测序峰图,并将每个样品的正反向序列进行拼接,得到的片段保存为fasta格式文件。将所得序列与NCBI中已经登记的基因片段进行BLAST相似性比对,初步判断确定扩增产物的可靠性。
种间遗传距离和种内遗传距离之间存在一个明显的差异,我们将这个差异称为DNA barcoding gap,这是DNA条码分析的一个重要部分。如果一个类群序列间存在着明显的barcoding gap,表示其种内的遗传距离明显小于种间遗传距离,那么它们就很容易被区分。相反,如果只有一个很小的或不存在DNA barcoding gap,那么这个类群的条形码物种界定就不清晰,利用DNA条形码去识别物种时就有一定困难。为了检验样本种间与种内个体间遗传变异的差异是否显著,我们使用MEGA X软件以K-2P模型计算COI基因片段的种内及种间遗传距离并进行统计,统计频率分布,进行barcoding gap分析。本项目中我们收集到了7科9属9种13个样本的鞘翅目害虫,利用这13个样本的COI基因片段和目前已发表的同属物种的COI基因片段(薪甲因未鉴定到物种,选择两个有记录的属),以属(或科)为单位,做了9个barcoding gap分析。
在数据的整理中,我们规定纳入分析的条形码数据要满足以下条件:保证所有条形码的同源性,因为发表的COI数据同源性并不统一,有的为该基因的前半段而有的为后半段,我们自己测序了COI基因的全长序列,可根据需要删减部分片段和序列;COI片段长度大于500bp[为了能够计算该种的种内遗传距离差异,锯谷盗属(Oryzaephilus)属中大眼锯谷盗(O.mercator)保留了3条500bp以下的数据];排除未鉴定到属的序列,即物种为某科的sp.的序列不在数据集中。
根据MEGA X中Distance模块的分析,将510条符合条件的数据根据科或者属分类(510条数据包括本实验测序的18条序列及数据库下载的492条序列,下载数据的数据库索引编码和分类信息详见附录),整理为9个组,每个组内计算出样本的种间遗传距离和种内遗传距离[除薪甲科(Latridiidae)之外,每个组中包括本次采集样本的同属物种的COI序列]。结果表明平均种内遗传距离为0.018(最大为0.053,最小为0),平均种间遗传距离为0.215(最大为0.333,最小为0.106),超过种间遗传距离11.9倍(表2)。依据Hebert等提出的利用COI序列进行物种鉴定的前提,种间遗传距离大于种内遗传距离差异,且一般种内遗传差异在2%以内,种间遗传差异至少是种内遗传差异的10倍[16] ,在理论上可以利用COI基因条形码对档案鞘翅目害虫进行初步鉴定,但是我们在针对每个物种的单独分析中,也发现了仅用COI片段条形码进行物种界定时存在的问题(图1)。

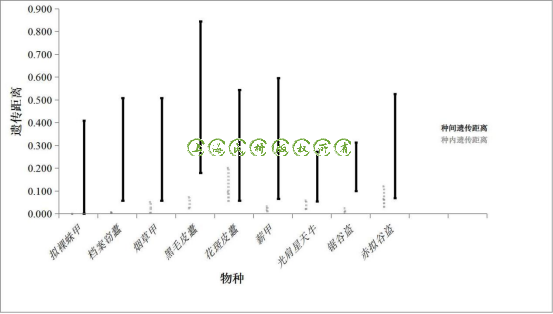
图1 分组的种间、种内遗传距离比较
以遗传距离的平均值±标准差来直观地看DNA barcoding gap,从图1中我们发现拟裸蛛甲、档案窃蠹、烟草甲、黑毛皮蠹、薪甲及锯谷盗都有明显的barcoding gap存在,但是花斑皮蠹、光肩星天牛和赤拟谷盗的种间遗传距离与种内遗传距离差异并不大,DNA barcoding gap不存在或不明显。因此我们以属为单位统计每个种的条形码数据的两两遗传距离差,比较种间遗传距离与种内遗传距离的分布频率(图2至图5)
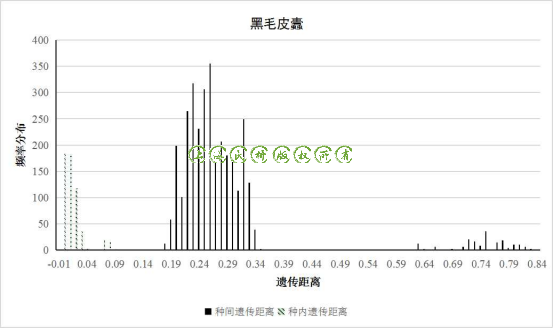
图2 黑毛皮蠹的种内、种间遗传距离频率分布
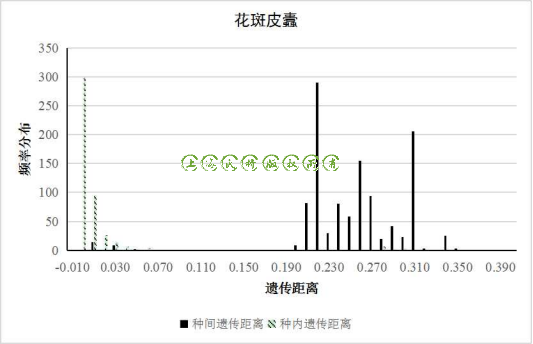
图3 花斑皮蠹的种内、种间遗传距离频率分布
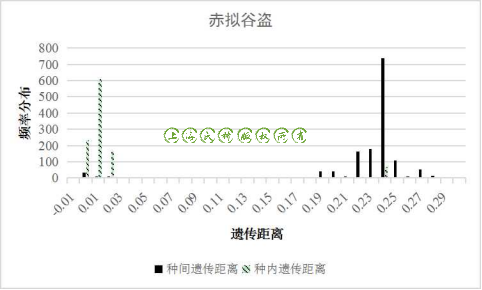
图4 赤拟谷盗的种内、种间遗传距离频率分布
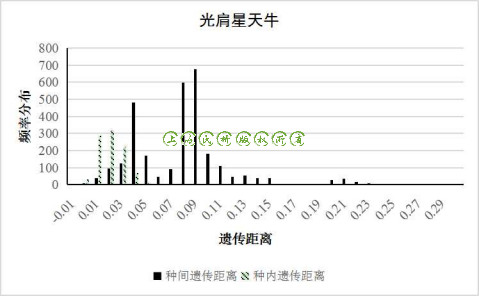
图5 光肩星天牛的种内、种间遗传距离频率分布
在遗传距离频率分布图中,我们可以发现黑毛皮蠹等物种的种内和种间的遗传距离分布没有交集(图2),而花斑皮蠹、赤拟谷盗和光肩星天牛的遗传距离分布出现了明显的交集,也正是这些交集掩盖了DNA barcoding gap(图3、图4、图5)。
因此我们进一步探寻其原因,发现在NCBI数据库中发表的数据中,HM398874.1_Trogoderma_parabile和HM398873.1_Trogoderma_parabile两条数据与花斑皮蠹的条形码数据遗传距离差异极小,可能是该数据是花斑皮蠹被错误的鉴定为斑皮蠹属的另外一个物种Trogoderma parabile,也可能Trogoderma parabile和花斑皮蠹这两个物种本身的COI基因差异很小,无法通过该条形码区分这两个物种;
KC440129.1_Tribolium_castaneum和JQ350711.1_Tribolium_castaneum与杂拟谷盗数据的遗传距离差异极小,可能是该数据被错误的鉴定为赤拟谷盗,从而掩盖了DNA barcoding gap。
星天牛属(Anoplophora)的数据情况较复杂,属内多个物种的种间遗传距离小于0.1,从而出现了明显的种间、种内遗传距离的交集,该属内共涉及12个物种,而这些物种相互之间均无法有效区分,即COI基因条形码片段在星天牛属的鉴定中很可能是无效的。这也解释了我们在整理数据时发现的该属COI基因条形码片段较长(70/90的片段超过900bp,而其他物种的有效条形码数据为600bp或更少)的原因,学者们可能尝试选用更长的序列片段作为星天牛的DNA条形码,以期更长的片段能够体现出更多的种间遗传差异。
综上,我们发现基于遗传距离的DNA barcoding gap分析能有效界定6个档案鞘翅目害虫物种,星天牛属的物种需要更多数据或者其他的方法才能进一步界定,而使用条形码数据时,原始数据鉴定的准确性直接决定了DNA条形码数据的效用,如果数据库中存在过多的鉴定错误的数据或者污染严重的不可靠数据,在利用数据库进行比对和遗传距离分析时,会出现很大的误差。
3.2 基于DNA条形码的系统发育树构建结果及分析
除了DNA barcoding gap的分析,我们还可以利用条形码数据进行系统发育分析,通过序列间亲缘关系的聚类分析来判断样本间的遗传距离差异是否正确体现了其亲缘关系,因此本项目利用510条COI基因条形码数据作为内群,用德国小蠊的同源COI基因片段做外群,在MEGA X软件的Phylogeny模块进行了邻接法(图6)建树,使用K-2P模型,自举值设为500。
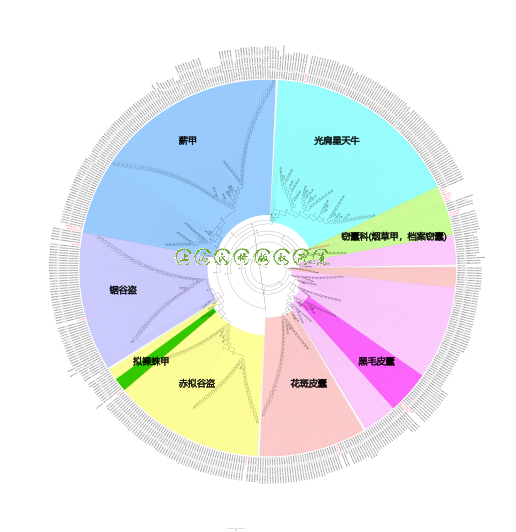
图6 基于COI基因条形码片段的档案鞘翅目害虫NJ系统发育树
根据系统发育树的拓扑结构,NJ法可以界定本次研究取样的绝大多数物种,即使星天牛属的物种个别为复系,但是拥有三条以上数据的种都可以聚为单系,对于同属的物种,系统发育树中无法有效构建斑皮蠹属和拟谷盗属的单系性(非单系,即没有完全聚类),这些属内部的物种分布在树的不同位置,而并没有聚为一支。系统发育分析能够更好的区分一些种内遗传距离较大的物种,比如花斑皮蠹和赤拟谷盗,虽然二者所在的属在树图中分布零散,但是两个物种本身的单系性得到的稳定的支持。
我们比较了基于系统发育树的界定方法和基于对比遗传距离的界定方法,两种方法均能有效的界定不同的物种,遗传距离分析中种内遗传距离偏大而无法有效界定的物种,原因可能是标本中存在其他形态相近的种或隐存种。基于建树的分析比基于遗传距离的界定更准确,而基于遗传距离的方法更加的简单高效,实际在评价某类群某种条形码的适用性时,需要结合两种方法进行比较和借鉴。
以黑毛皮蠹的成、幼虫COI基因条形码为例,本项目采样测序的成、幼虫的COI基因条形码序列与已公布的同物种条形码序列比对时,在645bp的序列上仅出现了13个碱基的差异,其余的位点均完全一致,表明黑毛皮蠹不论是在成、幼虫之间,还是在世界不同种群之间,都保持了很高程度的基因保守性。因此,利用COI基因作为条形码序列,可以很高效的鉴别形态上难以辨认的幼虫、若虫、蛹、甚至虫体残躯。基于同样的原理,DNA条形码目前在食品行业中多用于鉴定食材来源,如鱼翅、肉干等海鲜类产品和调理肉制品。
为了提高鉴定的精准度,我们认为可以增加测试片段,除了COI基因,还可以添加18S、28S等不同的基因 [17],用多个片段建立数据集,其效果和在用形态数据进行系统发育分析中增添多个有效辨别特征一样,分析结果会更可靠。当测试片段足够多的时候,可以找到不同种之间最有效的鉴定条形码,确定实际检测中利用不同的条形码来界定物种。
通过本次研究,我们认为DNA条形码物种界定的技术应该建立在物种信息准确、片段数据详实的数据库的基础上,然后通过分析样本的条形码数据找出样本和数据库分子数据上的异同,判断分子数据的差异性和形态差异性是否一致,决定是否需要更多的分子片段才能完全体现出种间差异。对于形态上差距甚小的“隐存种”,或者鉴定形态不分明的正在进化中的种,大量的分子数据可能比形态数据更有说服力。对于形态上无法区分的样本,可以先分析其分子数据,尤其对于分子数据差异较大的样本,可建立更大的数据集做更深入的分析。
我们在分析数据时,发现了一些分子数据种内的差异较大,尤其是对于常见的、广布的、基因交流频繁的黑毛皮蠹、花斑皮蠹、烟草甲、赤拟谷盗等,这些常见的仓储物害虫随货随粮随人传播的可能性更大,不同的采集地点得到的同种的样本很可能存在稳定的遗传差异,因此要注意这些物种的种内遗传差异,而对于毛皮蠹中亚种较多的物种,更需注意种下遗传距离的区分。相反,如果COI基因片段不能体现某些物种间的差异或某些明显的稳定的形态差异(如本研究中的星天牛),一方面要考虑到同种异型的现象(如百怪皮蠹),另一方面也要考虑控制某形态差异的基因不是COI,有必要进一步扩增更多的片段进行分析,若基因组数据无法得到结果,则可以进一步考虑进行转录组甚至代谢组的分析,因为动物形态的变化有赖于其基因表达过程中转录的差异以及代谢过程的差异。
本文系财政部基本科研业务费项目“档案库房鞘翅目害虫DNA条形码鉴定”(2022-3J-01)的阶段性研究成果。







 售前支持
售前支持